
【PRESS RELEASE】Expanding Molecular Dynamics Calculations Supporting Materials Development and Drug Discovery onto Quantum Computing Platforms
- Jun 1
- 7 min read
Updated: Jun 5
~ Quemix and DENSO jointly develop a new computational framework
that directly evolves distribution functions over time, along with NVT quantum circuits,
and successfully demonstrate a proof of concept for integration with quantum chemistry calculations ~
June 1, 2026

Quemix Inc. (“Quemix”; Head Office: Nihonbashi, Chuo-ku, Tokyo; CEO: Yu-ichiro Matsushita), a company engaged in the research and development of quantum computing algorithms and software, has developed core technologies for executing Molecular Dynamics (MD) simulations*1 on quantum computers through joint research with DENSO Corporation (“DENSO”; Head Office: Kariya, Aichi; President & CEO: Shinnosuke Hayashi).
MD is an essential computational technology across a wide range of industrial fields, including materials development and drug discovery, where atomic-scale behavior directly influences product performance. Conventional MD methods are known to face limitations in accurately reproducing time evolution and spatial scales close to real-world environments.
In this research, the two companies proposed a “quantum-classical hybrid MD framework” that combines quantum and classical computers for MD calculations and successfully achieved low-cost and highly accurate prediction of chemical states.
This approach demonstrates a pathway toward advanced utilization of quantum computers in fields requiring precise atomic-level analysis and functional property prediction, including next-generation battery materials, polymer materials, catalysts, and drug discovery.
Going forward, the companies will continue improving and expanding this technology while pursuing industrial applications in preparation for the era of Fault-Tolerant Quantum Computers (FTQC).
Through this joint research, the two companies successfully reformulated Molecular Dynamics (MD) into a new computational framework in which distribution functions*2 representing the spatial and momentum distributions of atoms are directly maintained and time-evolved as quantum states. In conventional MD, constructing distribution functions requires tracking the motions of large numbers of atoms over extended periods of time and calculating physical properties based on the resulting ensemble of trajectories. In contrast, the newly developed method enables the distribution function itself — representing the ensemble of trajectories — to be directly represented and manipulated as a quantum state. This allows physical properties such as diffusion coefficients to be calculated more directly and efficiently.
In addition, the researchers presented a method for implementing constant-temperature (NVT) conditions, which are essential for reproducing real-world environments, on quantum circuits. They also established protocols for efficiently extracting physical properties, including diffusion coefficients and vibrational density of states, directly from quantum states.
Furthermore, the developed quantum-classical hybrid MD framework was applied to chemical-state calculations for hydrogen molecules (H₂), demonstrating that the method can be executed on quantum computers.
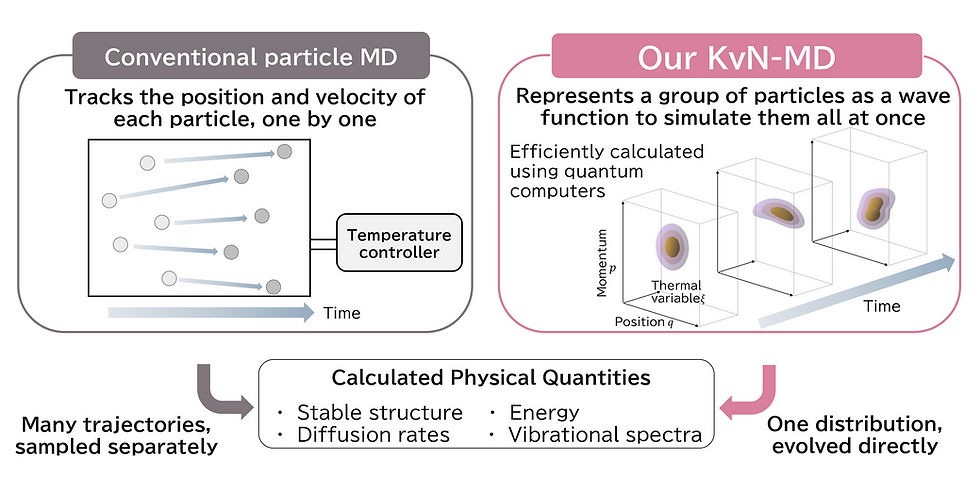
Figure 1: Schematic illustration showing the difference between the particle-based method used in conventional molecular dynamics simulations and the Koopman–von Neumann (KvN) approach.
Challenges Faced by Molecular Dynamics Simulations
Molecular Dynamics (MD) simulations are a foundational technology for manufacturing and scientific research, in which the motions of atoms and molecules are sequentially tracked based on Newton’s equations of motion. MD connects nanoscale phenomena with macroscopic material properties relevant to everyday life — including material strength, thermal conductivity, and gas permeability. MD is widely used across industrial research and development in areas such as extending battery lifetime, designing functional polymer and soft materials, predicting protein-drug interactions in drug discovery, and optimizing chemical processes.
At the same time, conventional MD faces major challenges associated with computational cost. Macroscopic physical properties such as diffusion coefficients and transport coefficients are obtained by tracking the motion of systems composed of large numbers of atoms over long periods and evaluating time averages and time-correlation functions along the resulting trajectories. Consequently, achieving higher accuracy generally requires dramatically increased computational time and larger simulation scales.
Although the potential use of quantum computers for MD has long been discussed, several technical challenges have been identified in constructing practical computational frameworks. These include:
representing classical distribution functions as quantum states
handling temperature and thermal bath conditions
efficiently extracting physical quantities from quantum states
These issues have remained key challenges toward the practical realization of molecular dynamics calculations using quantum computers.
Overview of the New Technology: Direct Time Evolution of Distribution Functions
To address these challenges, the two companies reformulated MD using a framework known as the Koopman–von Neumann (KvN) formalism*3. The KvN formalism is a mathematical framework that describes the motion of classical particles as the time evolution of wavefunctions, analogous to quantum mechanics, and can schematically be represented by the following equation:
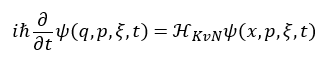
In this framework, |ψ|² corresponds to the distribution function. In conventional MD, distribution functions are indirectly evaluated through long-time trajectory tracking and statistical averaging. In contrast, the proposed method enables the distribution function itself to be directly maintained and manipulated as a quantum state. This provides a foundation for new computational methodologies that leverage the intrinsic characteristics of quantum computers.
Based on this technological foundation, the two companies developed the following technologies:
Quantum-Circuit Implementation of Temperature-Controlled (Nosé–Hoover NVT) MD
The researchers reformulated the Nosé–Hoover NVT method*4 — a deterministic thermal bath widely used in MD simulations — into a wavefunction representation compatible with quantum circuits and demonstrated concrete procedures for constructing the corresponding quantum circuits. This opens a pathway for handling simulations under “realistic conditions” on quantum computers.
Extension to Probabilistic Thermal Baths (Langevin NVT)
For Langevin-type thermal baths incorporating friction and stochastic fluctuations, the researchers demonstrated that implementation on quantum circuits is also possible by applying the Probabilistic Imaginary-Time Evolution (PITE®)*5 method developed and patented by Quemix. The ability to handle both deterministic and probabilistic thermal baths within the same framework significantly broadens the flexibility and applicability of the approach.
Efficient Extraction of Physical Properties The researchers established methods for extracting physical properties characterizing materials — including diffusion coefficients*6 and vibrational density of states (VDOS)*7 — from the time-evolved distribution functions. While measuring quantum states still requires repeated sampling, these physical properties can be efficiently calculated from quantum states by appropriately utilizing Quantum Phase Estimation (QPE)*8 and Quantum Amplitude Estimation (QAE)*9 depending on the target application.
Through these developments, the companies established a comprehensive quantum-computing scheme for MD calculations, covering the entire process from mathematical formulation and temperature control to the extraction of simulation results.
Completion of a Proof of Concept for a Quantum-Classical Hybrid MD Framework for Chemical-State Prediction
The two companies connected the newly developed quantum-classical hybrid MD framework within a single quantum circuit and performed chemical-state prediction calculations simulating the dissociation of hydrogen molecules (H₂) into two hydrogen atoms (2H) under high-temperature conditions.Conventionally, chemical-state prediction problems require computationally intensive frameworks combining MD with first-principles electronic-state calculations. As shown in Figure 2, the obtained results demonstrated that the developed framework enables highly accurate chemical-state prediction with substantially reduced computational cost.Although this achievement represents a proof of concept targeting simplified chemical-state prediction, it demonstrates the minimum framework required to enable chemical-state prediction on quantum computers. Applying the developed framework to more complex systems is expected to become feasible as the number of available qubits increases. Accordingly, this research provides an important pathway toward practical quantum-computing applications in the era of Fault-Tolerant Quantum Computers (FTQC).

Figure 2: Comparison between the conventional method and the newly proposed KvN-based approach. Rare-event sampling of H₂ → H + H dissociation at T = 5000 K.
(Left) Conventional method: Distribution function reconstructed from 1,000,000 trajectories initiated from the equilibrium bond length with thermal velocities.
(Right) Proposed method: Distribution function represented directly as a quantum state using the KvN formalism.
The results of this joint research are scheduled to be presented by researchers from Quemix and DENSO at “Q2B 2026 Tokyo,” an international conference on quantum technologies to be held at the Grand Hyatt Tokyo on June 4–5, 2026.
Q2B 2026 Tokyo Official Website: https://q2b.qcware.com/conference/2026-tokyo
Glossary
*1 Molecular Dynamics (MD) Simulation:A computational method for predicting material properties by treating atoms and molecules as particles governed by Newton’s equations of motion and tracking their time evolution on a computer. MD is a standard computational technology widely used in materials science, chemistry, and life sciences.
*2 Distribution Function:A function describing how the positions and momenta of particles constituting a system are distributed. Macroscopic physical properties can be calculated using distribution functions.
*3 Koopman–von Neumann (KvN) Formalism:A mathematical framework that describes classical mechanics in the form of wavefunction time-evolution equations analogous to those used in quantum mechanics. It serves as a bridge for naturally mapping classical systems onto quantum computers.
*4 Nosé–Hoover NVT:A deterministic thermal bath algorithm most widely used in MD simulations to maintain a constant system temperature.
*5 Probabilistic Imaginary-Time Evolution (PITE®):An algorithm developed and patented by Quemix for performing ground-state calculations and non-unitary operations on quantum computers.
*6 Diffusion Coefficient:A physical property representing how easily particles move within a material. It is a fundamental indicator directly affecting product performance, including battery charge-discharge characteristics.
*7 Vibrational Density of States (VDOS):A function describing how atomic vibrations in a material are distributed across frequencies. It provides fundamental data for calculating physical properties such as specific heat, thermal conductivity, and thermal transport characteristics, and is widely used in solid-state physics and materials engineering.
*8 Quantum Phase Estimation (QPE):A fundamental quantum algorithm for extracting information such as energy eigenvalues using quantum circuits.
*9 Quantum Amplitude Estimation (QAE):A quantum algorithm capable of estimating probability amplitudes contained in quantum states with fewer sampling trials than classical sampling methods.
About Quemix Inc.
Quemix Inc., a consolidated subsidiary of TerraSky Co., Ltd. (Headquarters: Chuo-ku, Tokyo; CEO: Hideya Sato), conducts research and development in quantum computing, quantum sensing, and computational materials science.
Under its vision of “realizing the future humanity has dreamed of through quantum technology,” Quemix supports breakthrough innovations for companies leading the next generation of quantum technologies.
Since its establishment in 2019, the company has focused on research and development of algorithms for fault-tolerant quantum computers (FTQC), including the development and patenting of the “Probabilistic Imaginary-Time Evolution (PITE®)” quantum chemistry algorithm, which has been mathematically proven to achieve quantum acceleration.
As a leading company in FTQC algorithm research in Japan, Quemix is actively advancing research and development aimed at the practical application of quantum computing in materials computation and simulation by 2030.
Contact Information
Quemix Inc.
Inquiry Form: https://www.quemix.com/en/contact





Comments